
Al menos una docena de productos médicos importados en el mercado argentino debieron ser retirados o corregidos por sus fabricantes extranjeros, a partir de que se detectaran fallas con potencial peligro de muerte para los pacientes. Entre ellos respiradores ydispositivos médicos para el corazón. Varios ingresaron al país incluso después que saltaran las alertas sanitarias en los Estados Unidos.
Esta información que hoy revela por primera vez en Argentina la investigación The Implant Files, del Consorcio Internacional de Periodistas de Investigación (ICIJ), hasta ahora no se conocía públicamente. Esto era así porque, según la Administración Nacional de Alimentos, Medicamentos y Tecnología Médica (ANMAT), los procedimientos llevados a cabo en cada caso son “confidenciales“.
La lista incluye desde dispositivos cardíacos y vasculares, hasta sistemas para asistir a los médicos durante las cirugías cardíacas o de cráneo, respiradores y equipos de diagnóstico por imágenes.
El equipo argentino de ICIJ – que integran Infobae, La Nación y Perfil– reconstruyó esta información a partir de cruzar la base de datos públicos de la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) que fueron sistematizados por ICIJ, con los registros de importaciones de la Aduana argentina, la información aportada por la ANMAT y las respuestas de las empresas importadoras a las consultas de los periodistas argentinos.
En Argentina no hay cifras oficiales sobre la cantidad de prótesis y dispositivos médicos que debieron ser retirados o corregidos en el mercado, hospitales e instituciones. En el área de Tecnovigilancia de la ANMAT, que se ocupa de los efectos adversos en los Productos Médicos (PM) autorizados en el país, explicaron que “están trabajando en estadísticas sobre los efectos adversos reportados”.
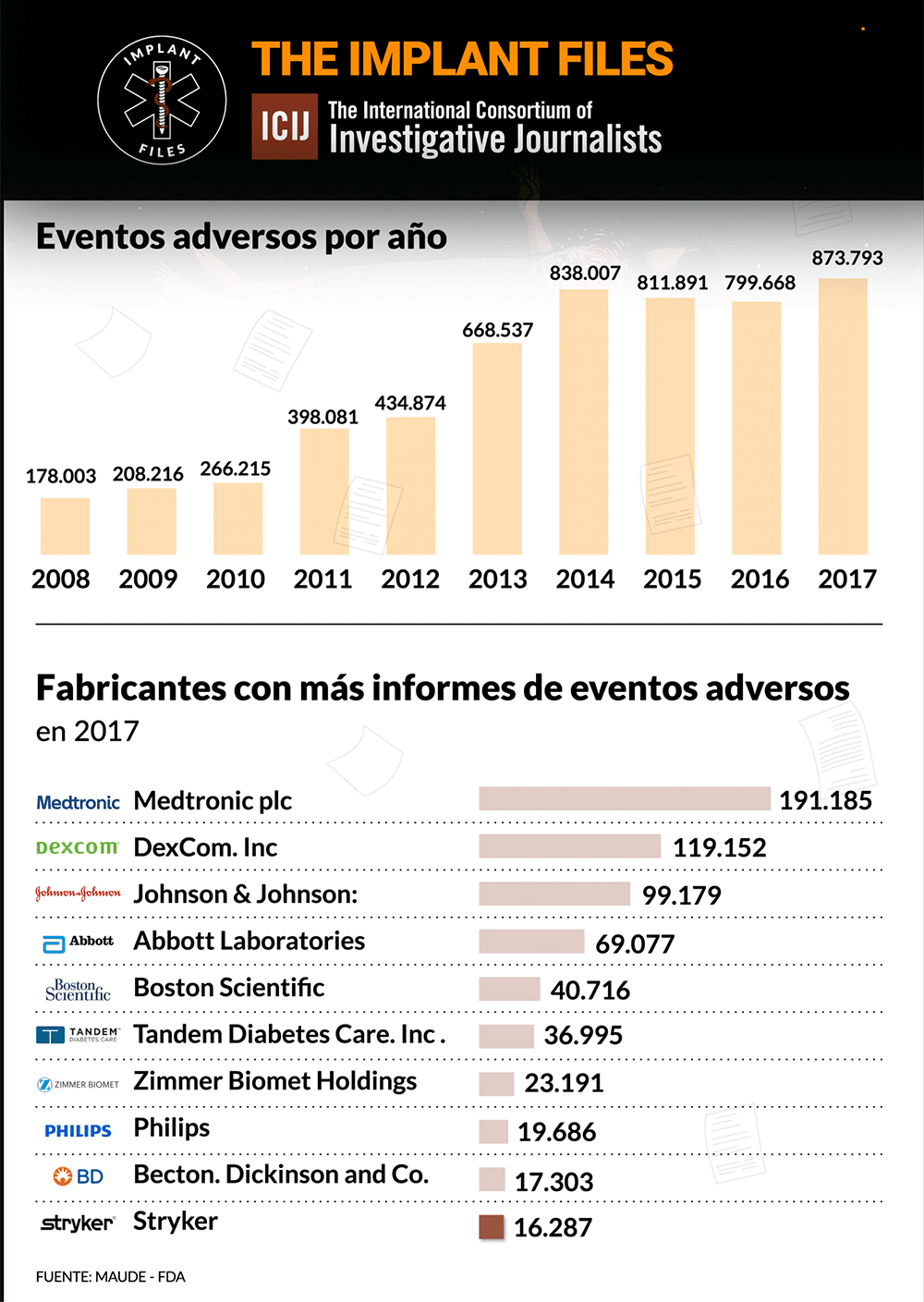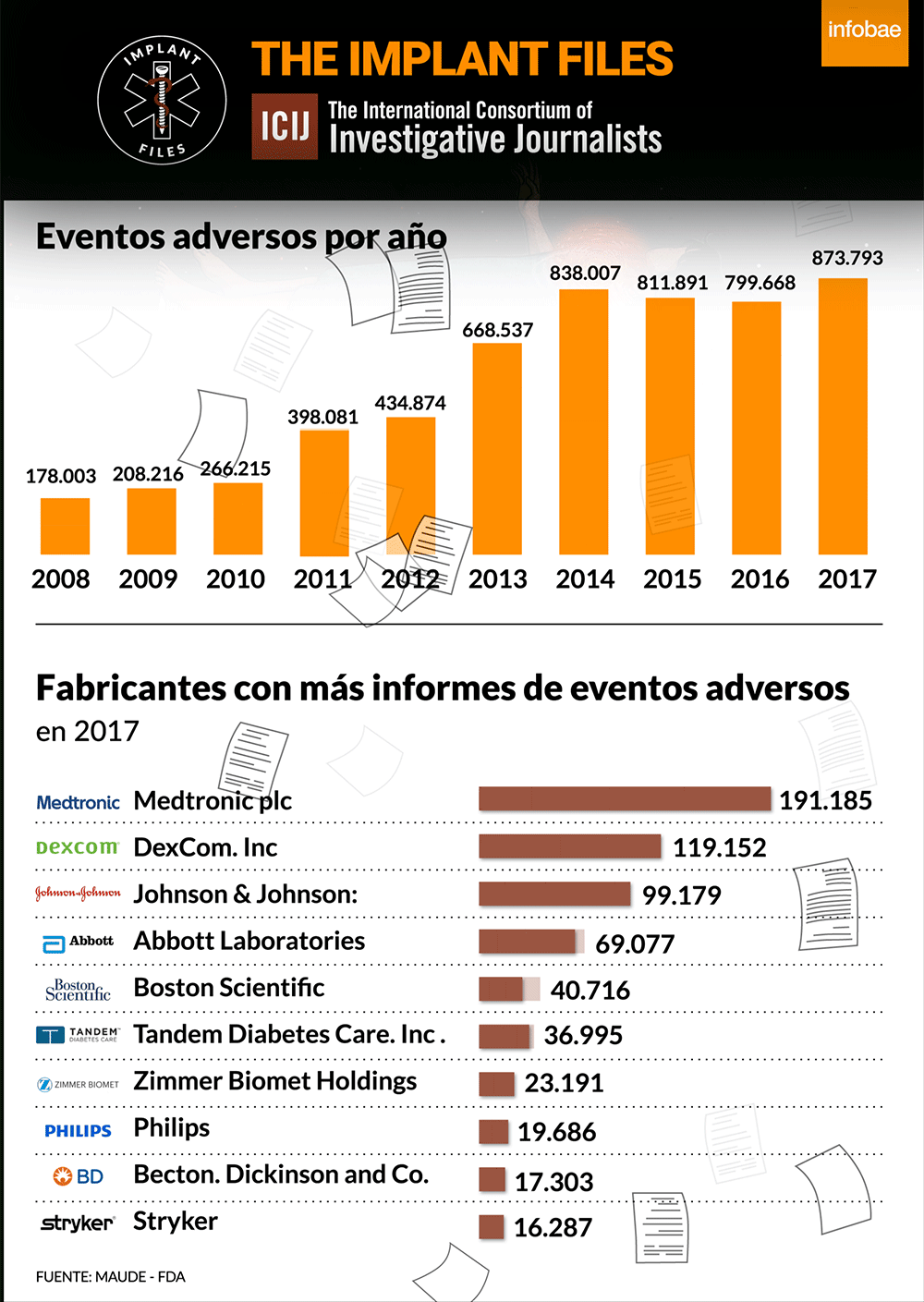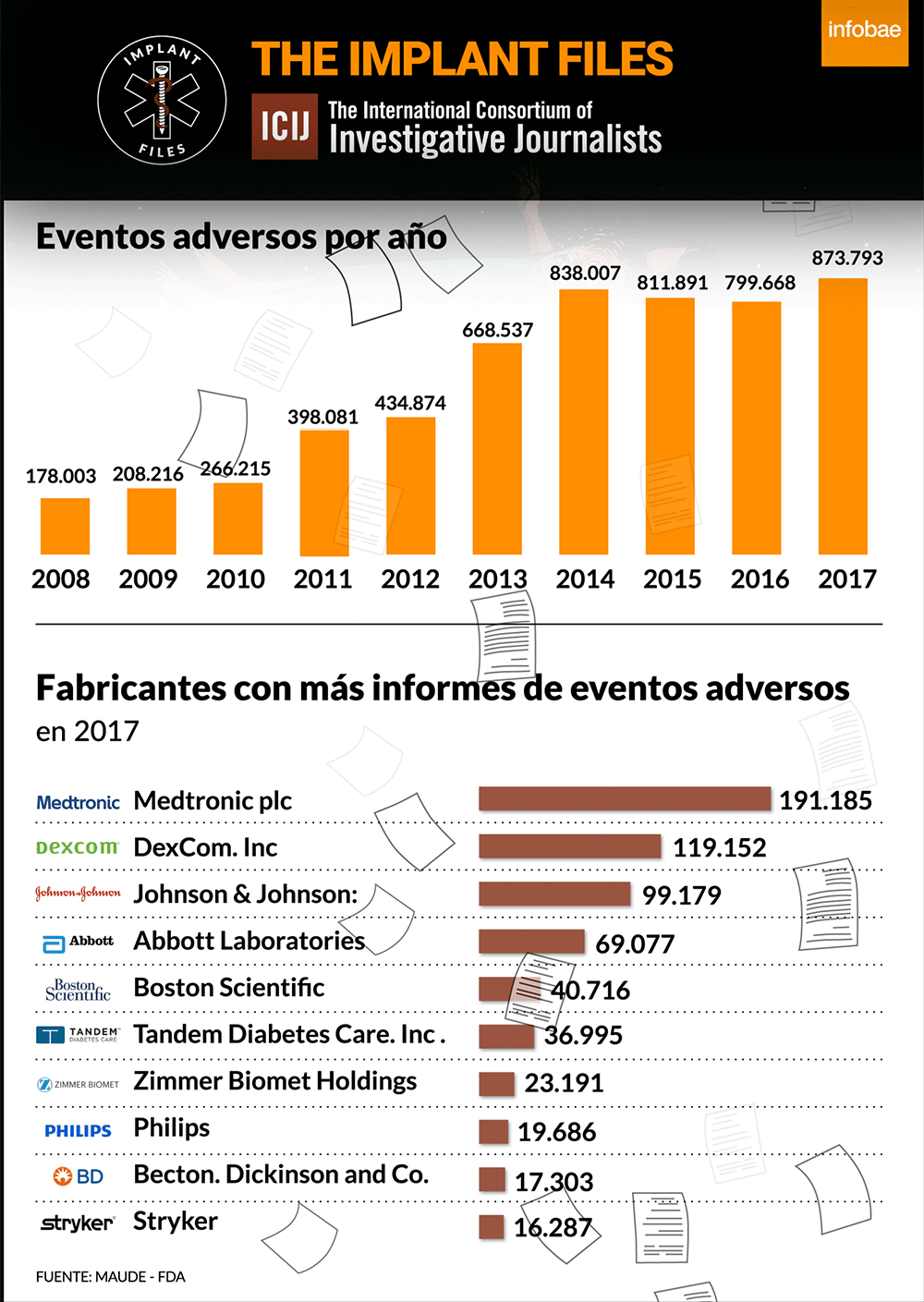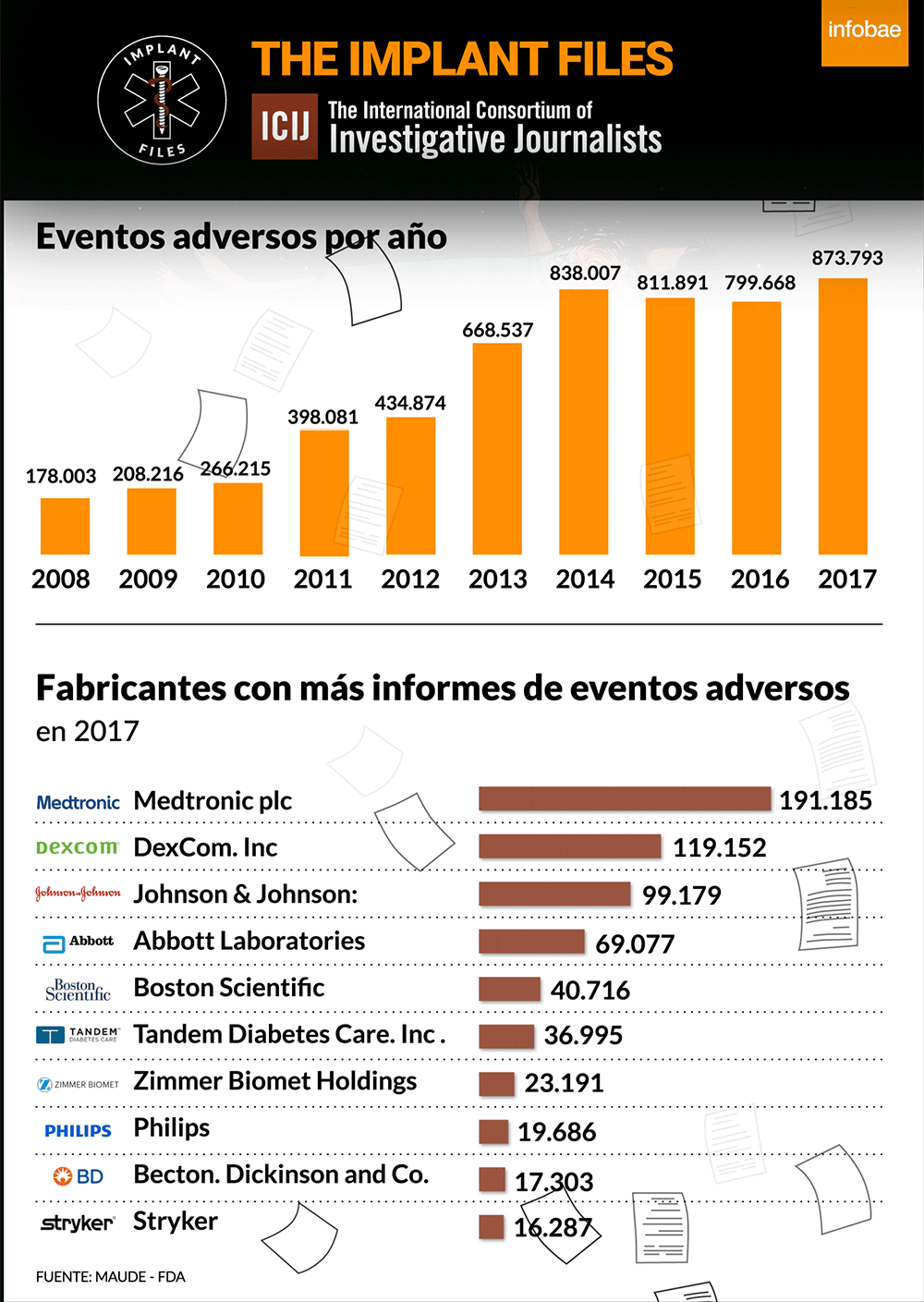
La FDA, la autoridad sanitaria de referencia en el mundo, publica diariamente lo que se denomina recalls o advertencias sobre distintos implantes y dispositivos médicos. Lo hace a partir de la información que brinda un fabricante ante la sospecha o certeza de que un producto puede tener una falla o provocar un riesgo a la salud. Según su gravedad, los califica de I a IV, siendo los de Clase I, los de mayor riesgo potencial, incluida la muerte.
Alertas con riesgo de muerte
El equipo argentino de ICIJ se concentró en identificar qué productos médicos con recall Clase 1 de la FDA se comercializaron en Argentina una vez emitido el alerta global. Para eso, revisó las autorizaciones de la ANMAT y buscó averiguar qué se hizo respecto de ellos una vez ingresados al mercado. Esa información – a diferencia de lo que sucede con la página web de la FDA- no está accesible en el sitio del organismo sanitario argentino.
Tampoco fue posible encontrar las disposiciones de la ANMAT publicadas en el Boletín Oficial con el aviso de retiro o la acción correctiva dispuesta sobre un implante o dispositivo, como sí sucede habitualmente con un medicamento, alimento o artículo cosmético considerados nocivos para la población.

Desde el organismo, sostuvieron que solo emiten una disposición pública “cuando hay una amenaza seria a la salud pública”. Explicaron que no lo hacen “si son casos muy puntuales de efectos adversos, para no alarmar a la población”.
También aclararon que un recall, no siempre significa el retiro del mercado del producto, sino que en ocasiones puede implicar que el dispositivo médico -si es un equipo par cirugía o de diagnóstico, por ejemplo- necesita ser revisado o arreglado. Cuando se trata de un implante ya colocado en el cuerpo -como una prótesis de cadera o malla quirúrgica, por ejemplo – no siempre puede ser extraído fácilmente de los pacientes. En ese caso, la fabricante delega en los médicos que se pongan en contacto con sus pacientes para decidir los pasos a seguir, en función del costo-beneficio de una nueva intervención.
Pese a la reticencia inicial de la ANMAT en dar precisiones sobre qué pasó en cada caso, finalmente ante la insistencia periodística, admitió que en la mayoría de los casos consultados las importadoras tuvieron que realizar alguna “acción de campo” correctiva o de retiro del producto ante el alerta sanitaria. Esta información fue corroborada por las compañías involucradas que accedieron a responder las consultas del equipo del ICIJ.
Entre los casos detectados figuran dispositivos cardiovasculares que, tras la alerta, tuvieron que parcialmente retirados y destruidos; bombas para el corazón que resultaron ser riesgosas y equipos de diagnóstico o cirugía que debieron ser revisados y reparados cuando ya estaban en funcionamiento.

El gigante Medtronic
Medtronic, una empresa estadounidense líder en dispositivos cardiovasculares, es una de que más retiros del mercado debió hacer en ese país y en el mundo, incluida la Argentina. Luego de que en los Estados Unidos, Medtronic informara que varios de sus productos tenían fallas que podían llegar a ocasionar la muerte de sus pacientes, esos mismos dispositivos destinados a personas con problemas cardíacos ingresaron y se distribuyeron en la Argentina.
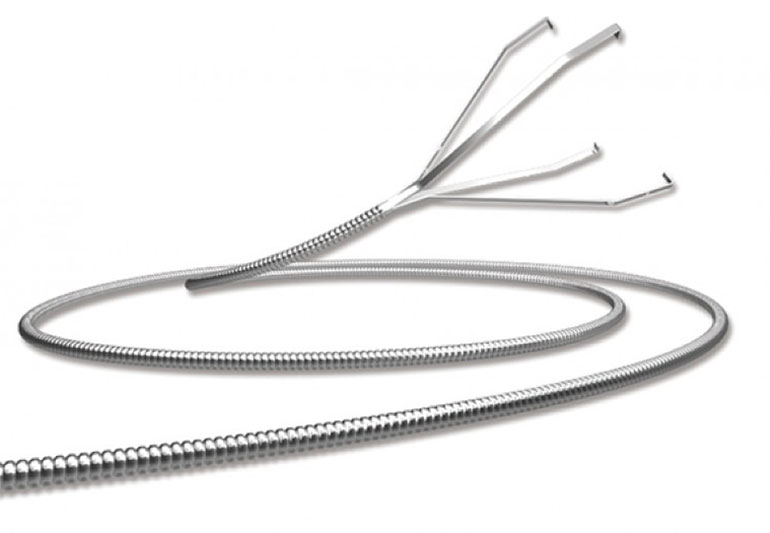
Uno de ellos es el dispositivo de recuperación Alligator Retriever, una pinza de platino diseñada para extraer cuerpos extraños de los vasos sensibles (venas, arterias y capilares). Se utiliza en casos de aneurisma y una herramienta quirúrgica desechable de un solo solo. El problema detectado por la empresa fue la deslaminación y desprendimiento del material de recubrimiento en el flujo sanguíneo del pacient, con riesgo de muerte.
La FDA emitió un primer alerta Clase 1 el 16 de abril de 2014 sobre 43 lotes, y un segundo aviso el 9 de noviembre de 2016 sobre otros 8 modelos del Alligator. Sin embargo, fue autorizado por la ANMAT para comercializarse en el país en abril de 2015.
Según los registros de Aduana, se importó el producto 18 días después del segundo alerta de la autoridades sanitarias de los Estados Unidos, y mientras estaba vigente. “Existió una acción de campo”, se limitó a responder la ANMAT sobre el dispositivo Alligator.
Ante la consulta del equipo argentino de ICIJ, Medtronic brindó más datos: “Todas las unidades que ingresaron a Argentina afectadas por el aviso de la FDA fueron importadas y vendidas por Biosud SA. El 7 de octubre de 2016, Biosud notificó a la Anmat sobre este recall y se comunicó con las instituciones de atención médica que habían adquirido el producto para solicitarles que lo devolvieran. Los dispositivos fueron destruidos. El distribuidor proporcionó evidencia de la destrucción el día 14 de febrero de 2017 a Medtronic y a la ANMAT”.
Desde Biosud, sin embargo, señalaron: “Nosotros ya no teníamos la representación de Medtronic cuando salió el recall. La acción le correspondía a Medtronic y colaboramos brindando información de los clientes”.

El Alligator no fue el único dispositivo que Medtronic debió retirar del mercado argentino. El 15 de noviembre de 2013 la FDA emitió una alerta Clase 1 sobre varios modelos de alambres guías orientables de Medtronic Vascular que se utilizan para colocar un catéter de ablación y permitir el mejor acceso al corazón del paciente durante la cirugía. Fue después que el gigante de la industria médica hubiera advertido que el recubrimiento de teflón se “descaramaba”, como las espinas de un pescado. El desperfecto podía causar la muerte de los pacientes.
La ANMAT había autorizado un año antes a Medtronic Latin America INC la importación de varios modelos de esos alambres guía que debieron ser retirados del mercado en los Estados Unidos: Zinger, Cougar Nitinol y Attain Hybrid.
Desde la autorización oficial y hasta junio de 2018 ingresaron a la Argentina más de 2000 unidades de distintos modelos, a pesar de la vigencia de la alerta sanitaria de los Estados Unidos. La Anmat sostuvo ante la consulta periodística que el importador “retiró el producto del mercado” argentino.
Desde Medtronic aseguraron que solo “40 unidades de este producto importadas a la Argentina resultaron afectados por este recall” y, según pudo sabe el equipo argentino de ICIJ, 34 unidades fueron devueltas a la empresa el 21 de noviembre de 2013 para ser destruidas. Otras 6 unidades ya habían sido utilizadas para el momento en el que llegó la alerta de la FDA. “No se recibieron observaciones o reclamos por parte de las instituciones de salud que las utilizaron”, dijo la empresa.
Un análisis de ICIJ de los informes de eventos adversos presentados ante los autoridades regulatorias de los Estados Unidos revela que Medtronic es la fabricante de dispositivos médicos con más efectos adversos reportados el año pasado: 191.185. De esta cifra, 1.828 se vincularon oficialmente a muertes, 71.444 a lesiones, y 117.901 a mal funcionamiento de los dispositivos.

